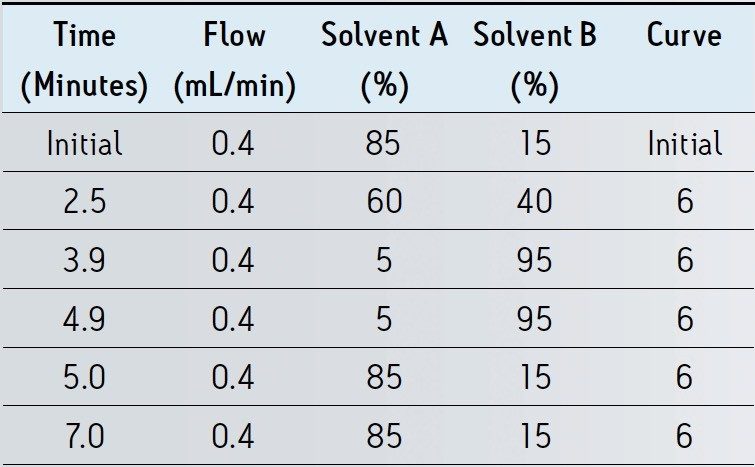
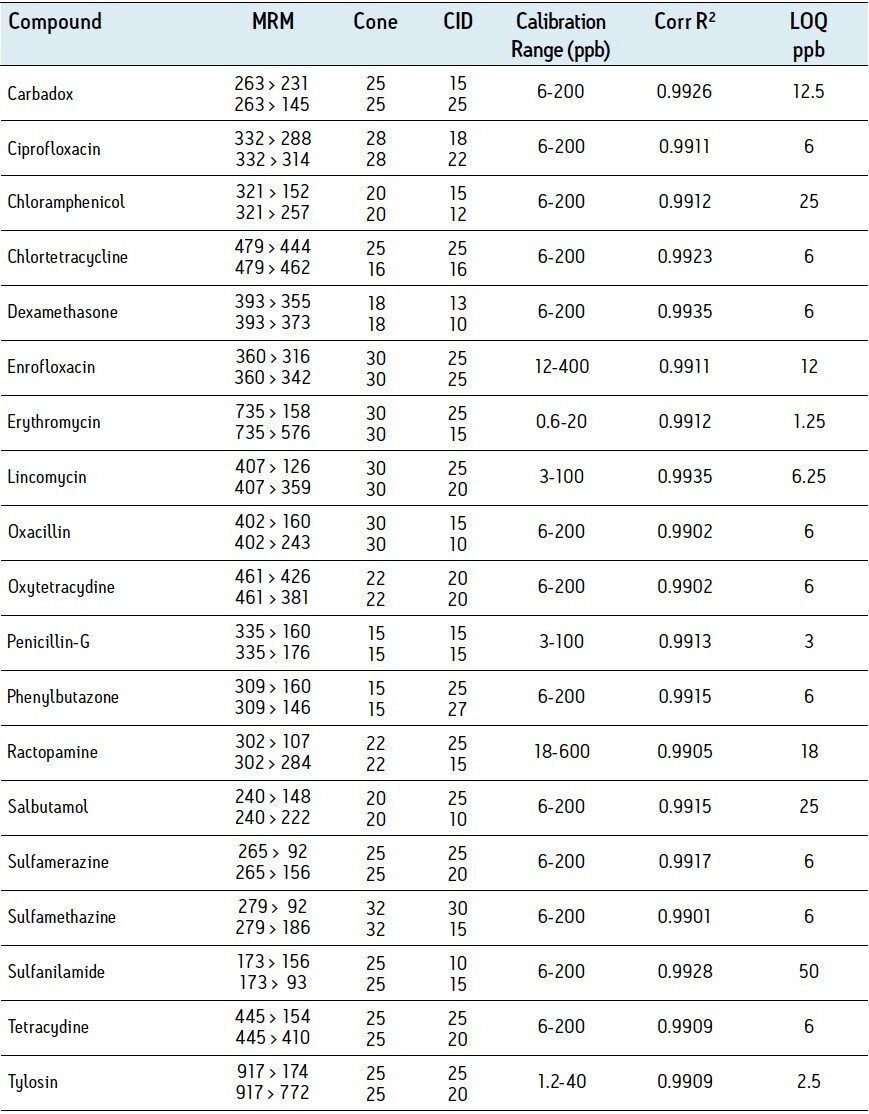
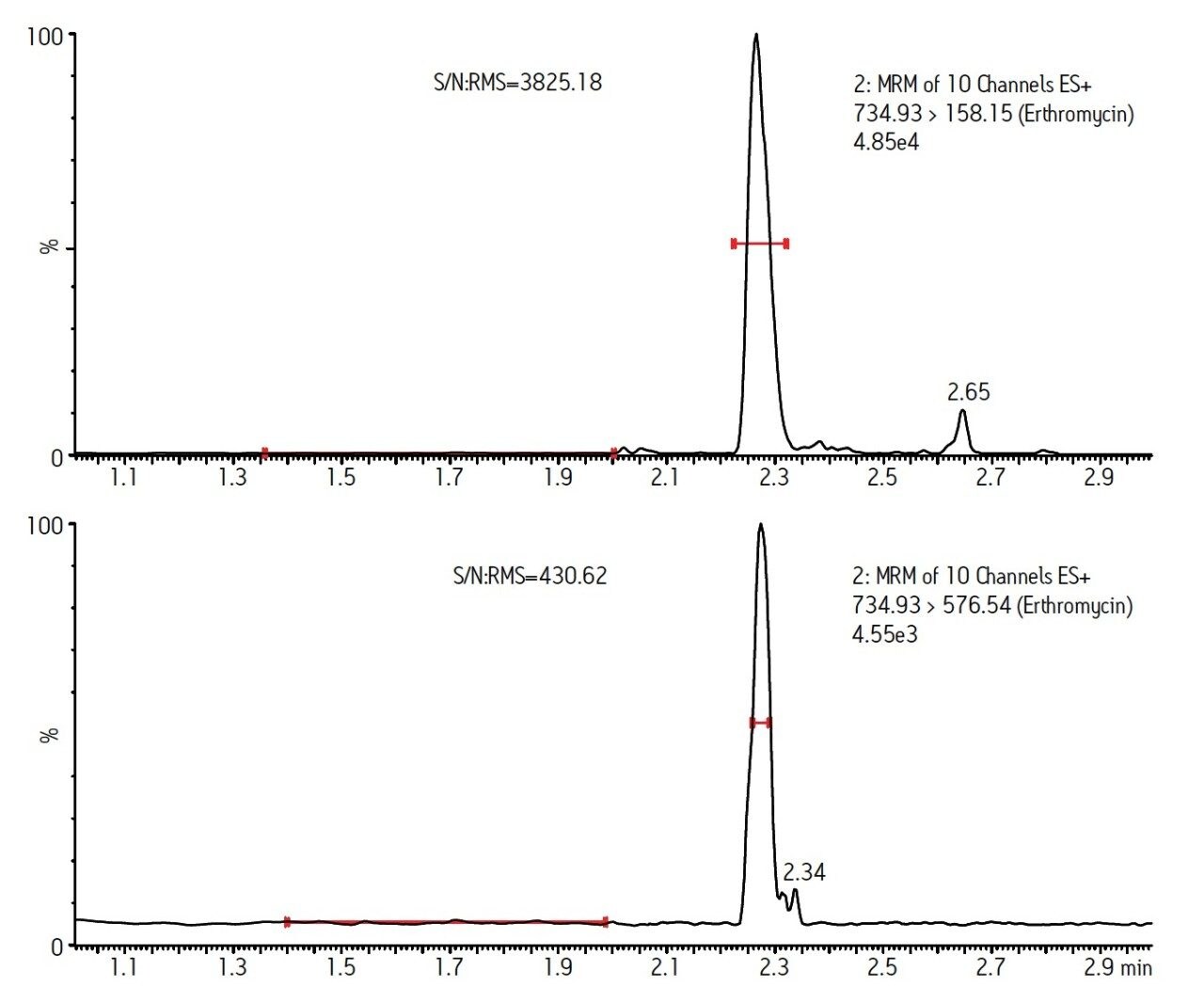
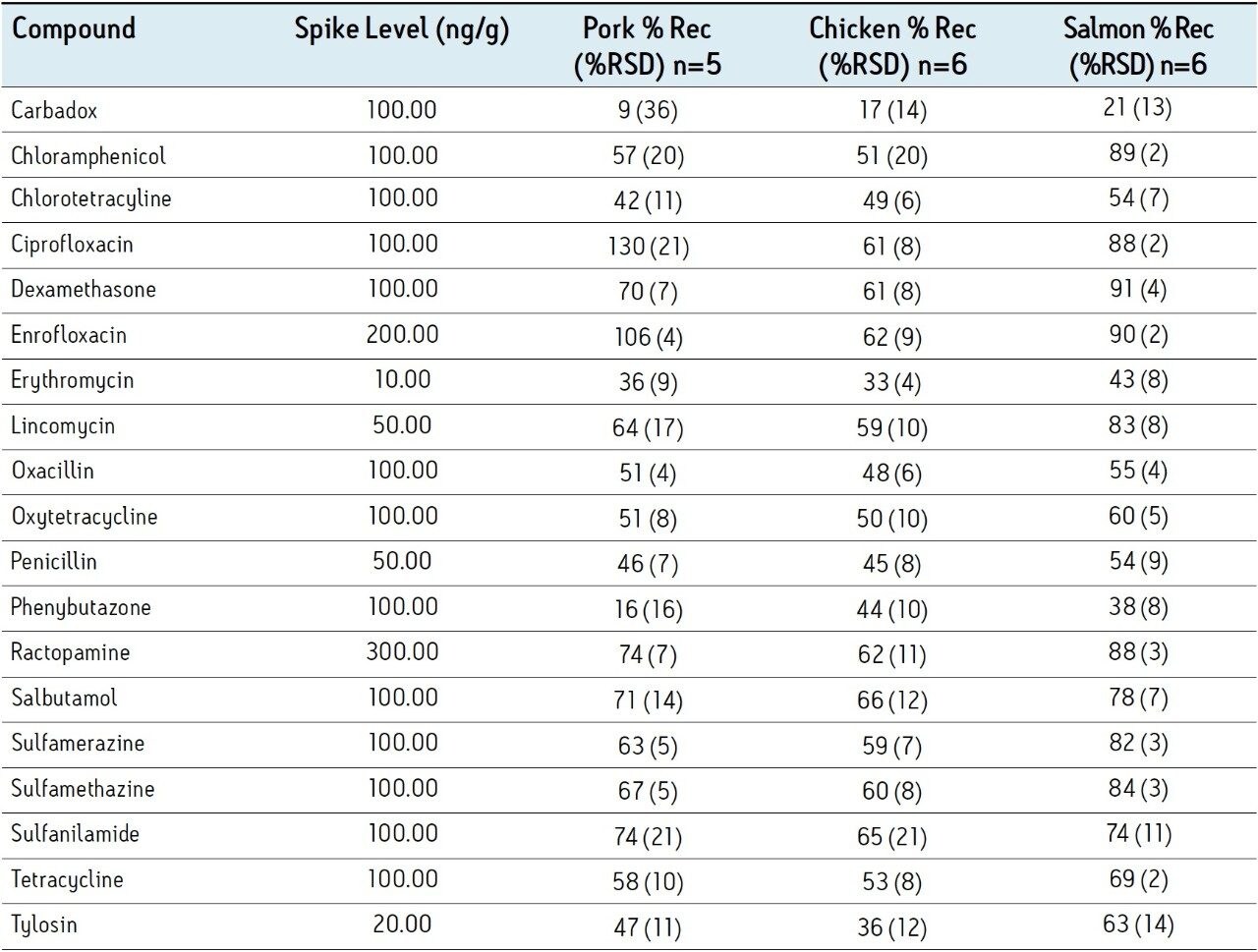
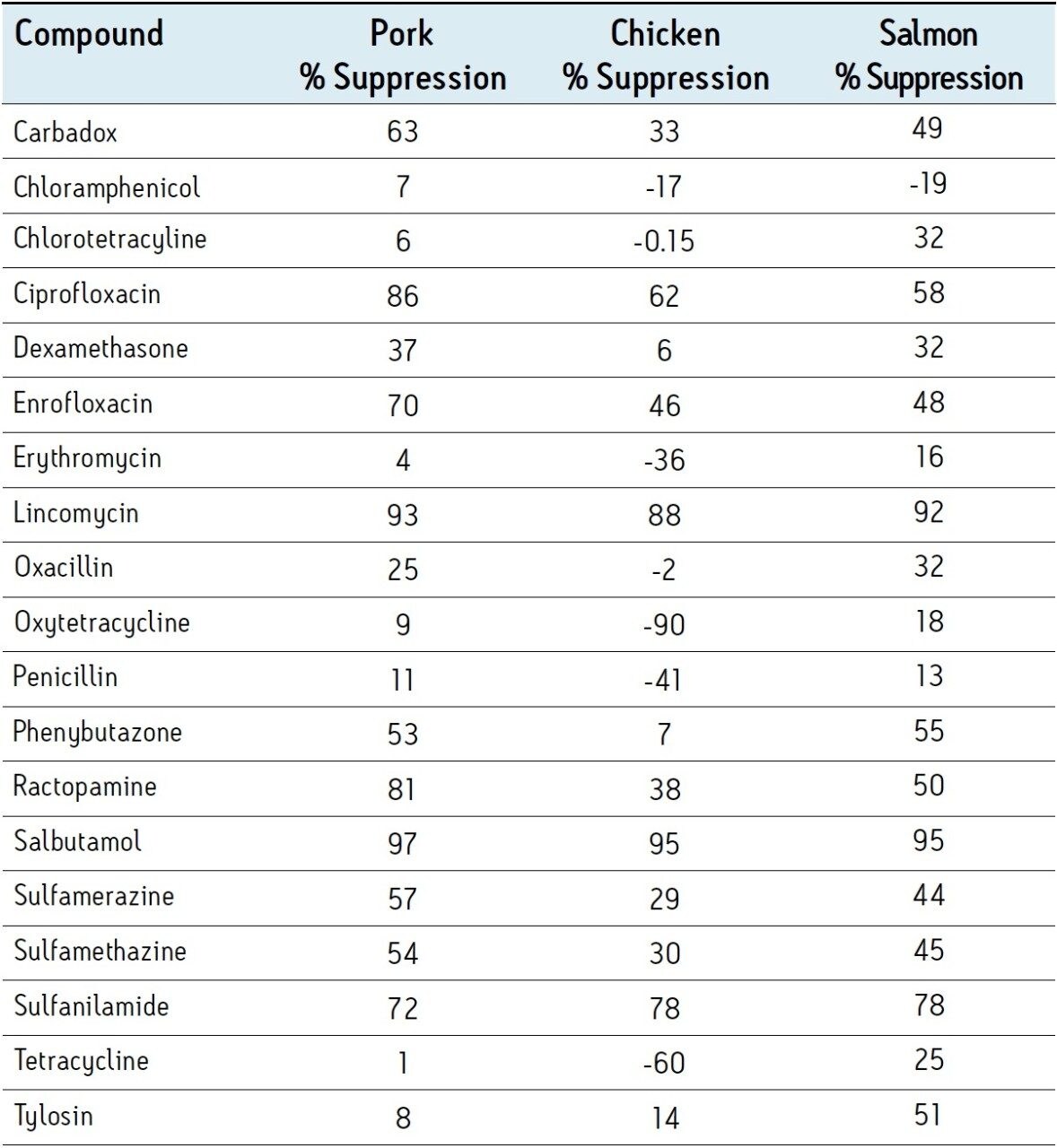
The procedure utilized in this study was developed from methods presented by Lehotay et.al.1 and Martos et. al.2. The method used in reference 1 uses no acid in the tissue extraction solvent; under these conditions we observed recovery of tetracyclines below 5% and the RSD for recovery of fluoroquinolones was greater than 50%. The method used in reference 2 prescribes the acidification of the extract to 1% formic acid prior to centrifugation; under these conditions penicillin recovery was under 10% compared with 48% using our approach. Our extraction procedure is a compromise of the methods presented in reference 1 and 2. A similar acetonitrile/water based extraction is used but is acidified only to 0.2% with formic acid; more balanced recovery and minimized degradation of labile compounds was achieved.
Another approach was considered based on the method of Kauffman et. al.3 by which two separate extractions were performed. The first extraction, for the water soluble compounds, was accomplished using aqueous succinic buffer. The second, performed on the re-suspended pellet, was with acetonitrile. This approach requires that each fraction be worked up independently before ultimately combining fractions for a single injection. Performance was only marginally better than the chosen procedure but at a much greater cost of time and materials. The extraction, cleanup, and analysis protocols chosen for this study provide a good balance of preparative time, cost and method performance.