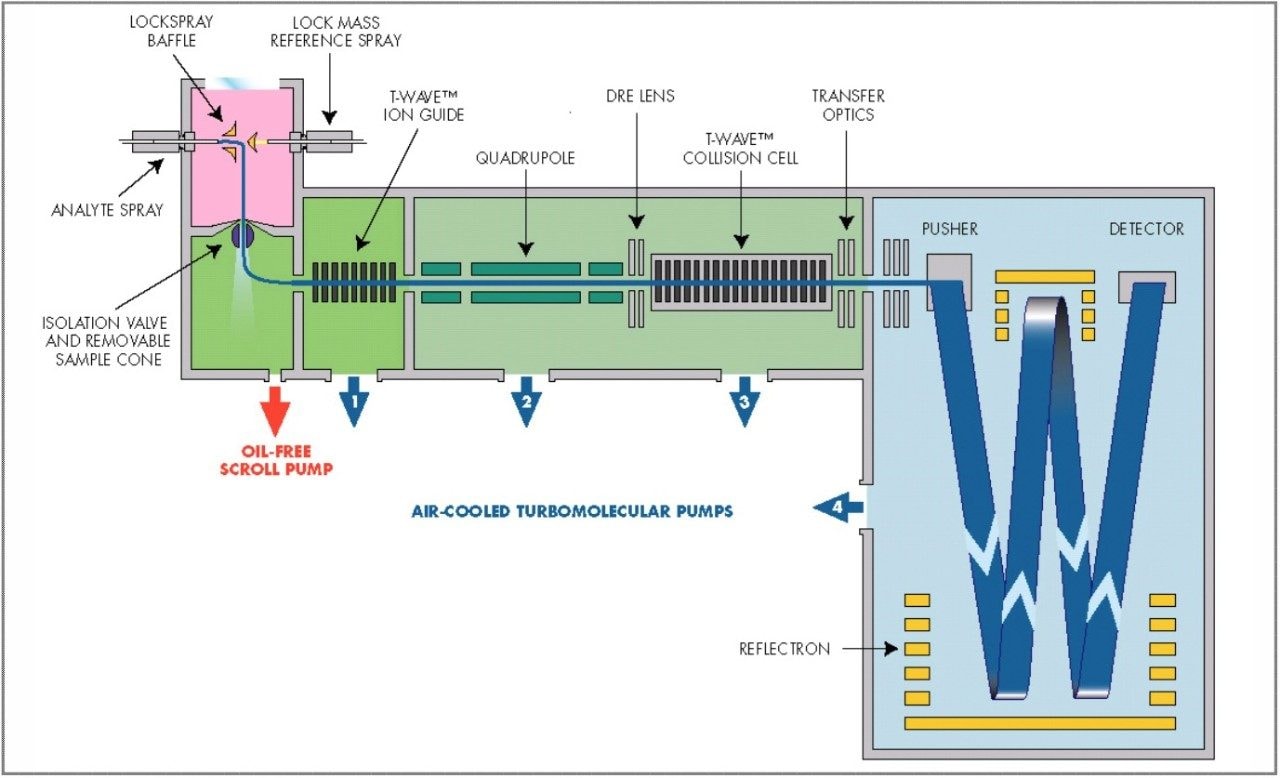
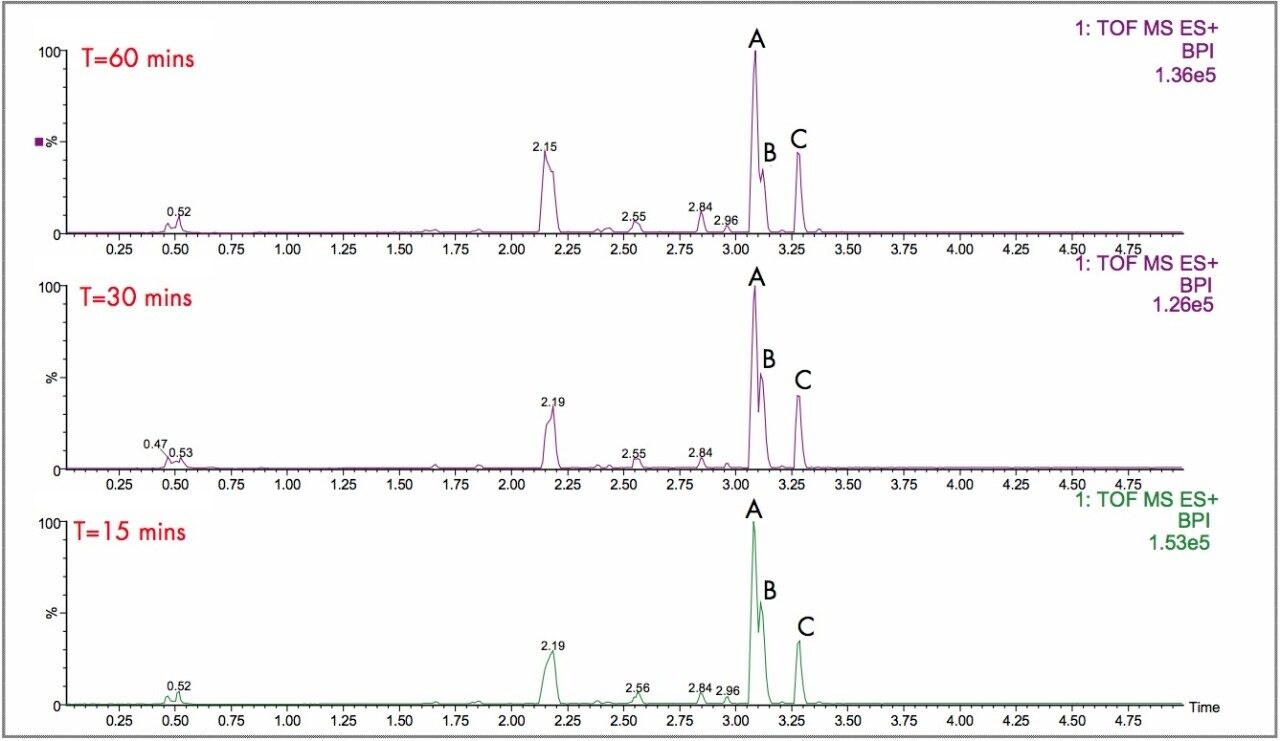
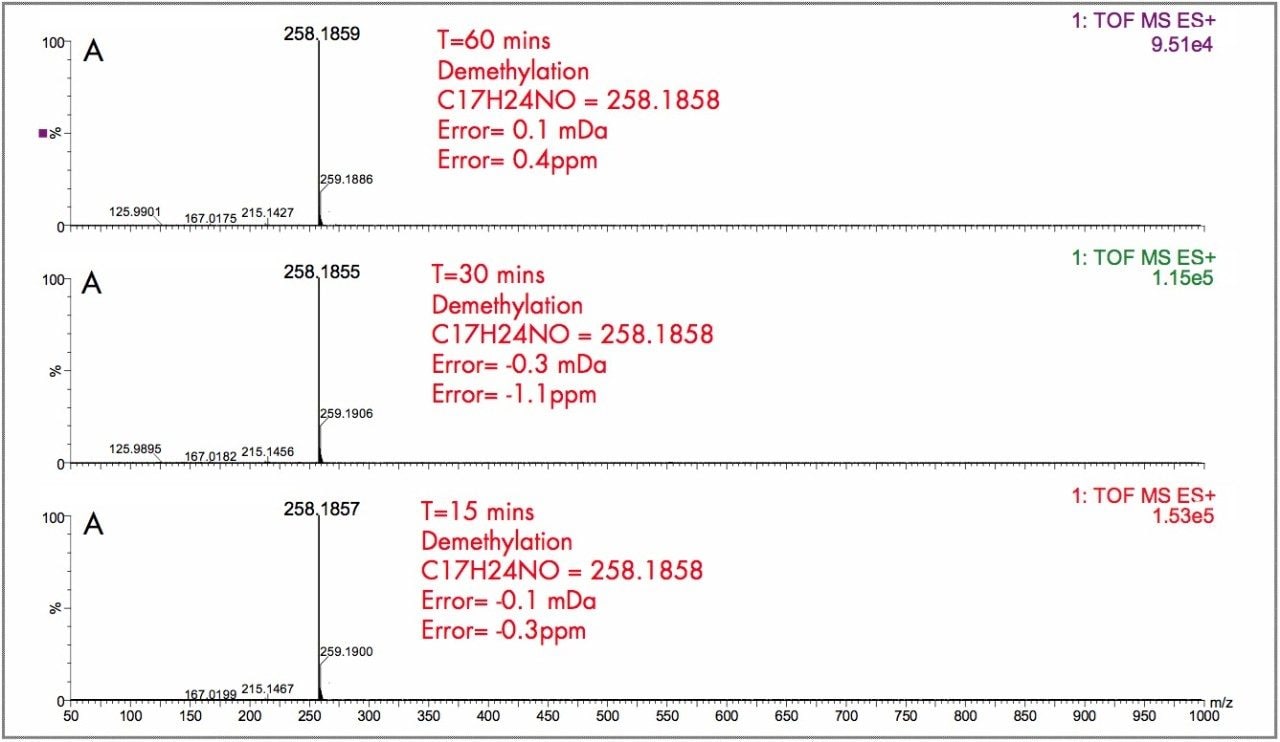
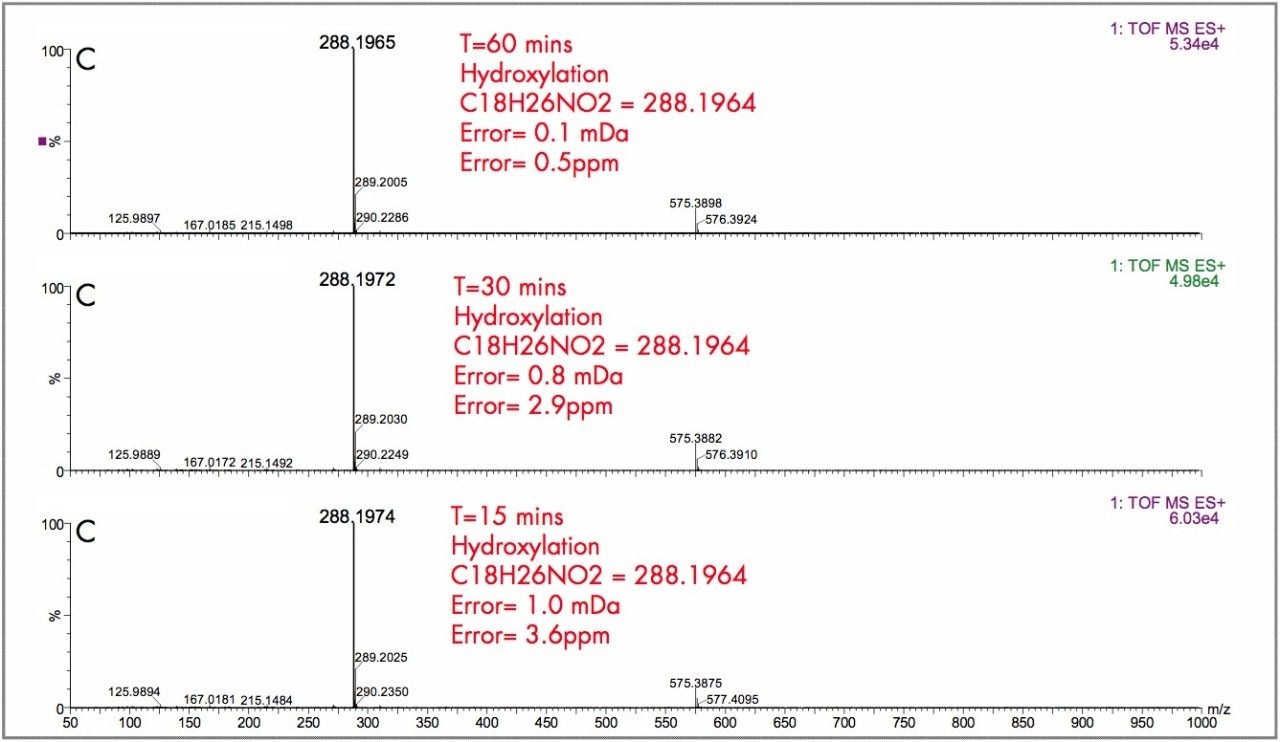
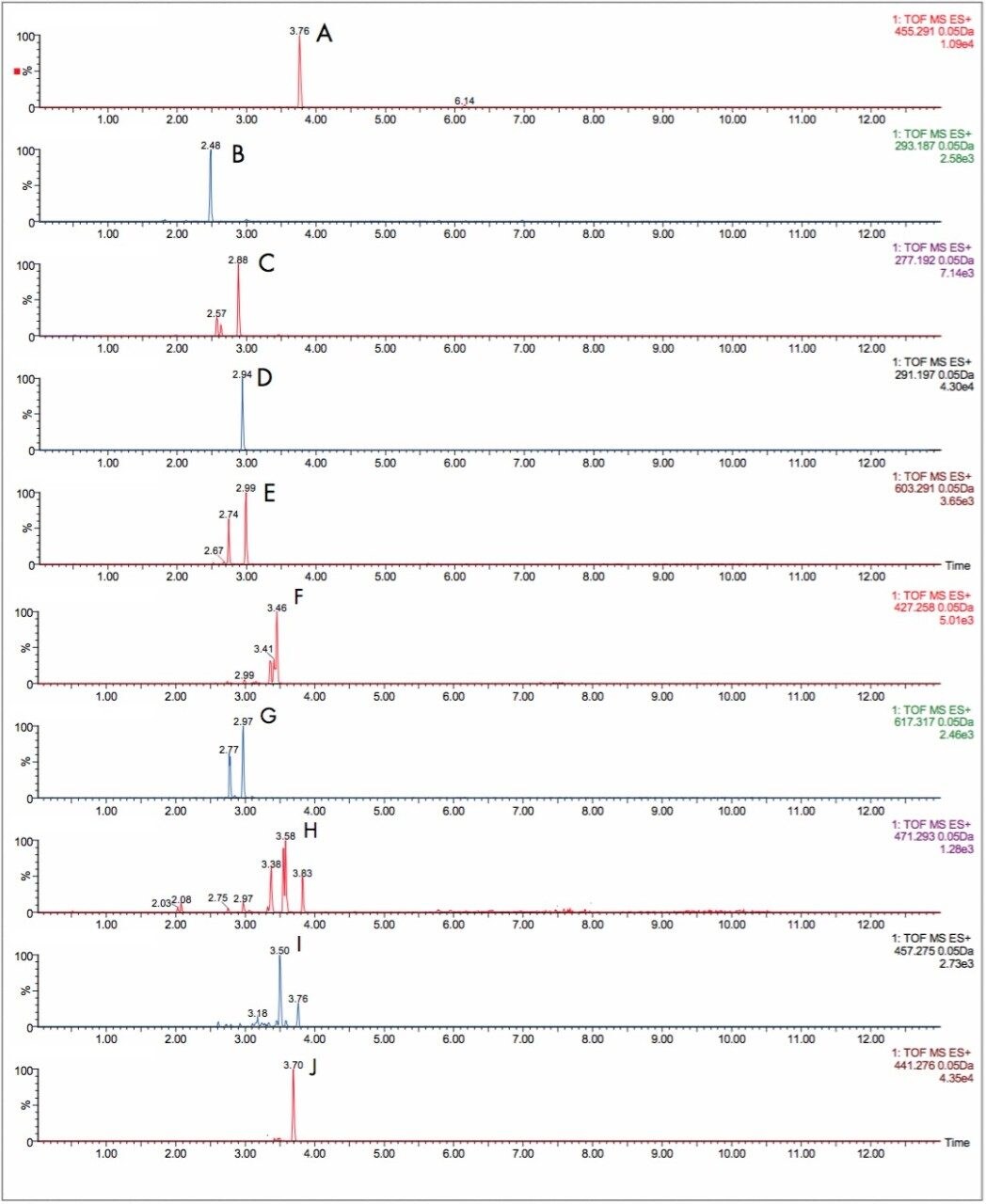
The parent drugs verapamil and dextromethorphan were incubated in vitrousing human S9 microsomes. These incubations were utilized to test both hardware and software enhancements for determining the metabolic fate of drugs. The schematic of a Q-Tof Premier in Figure 1 shows the pDRE lens, which enhances the analytical applicability of oa-Tof MS. This feature has enabled extended dynamic range up to 4 orders of magnitude, providing both qualitative and quantitative capabilities with good exact mass measurements for highly concentrated peaks. Previously, at high ion counts, a shift to lower mass would occur, giving incorrect mass measurement and less confidence in elemental composition determination. Selection of spectra at low ion counts would provide excellent mass measurement and high confidence in the resultant data, however, this could be a laborious task. Even at high ion counts, pDRE allows either the peak top, or the whole peak to be averaged to generate an exact mass spectrum. This improves the ease with which data can be generated and processed. Also, since no detector saturation is taking place, a true profile of the route and rate of metabolism will be obtained. The impact of dynamic range enhancement can be seen in Figure 2 where the UPLC-Tof BPI chromatograms formetabolites formed, for the in vitroincubation of dextromethorphan are shown at time points 15 min, 30 min, and 60 min. A true chromatographic profile is observed. The exact mass spectra obtained for the demethylated (labeled “A”) and hydroxylated metabolites (labeled “C”), can be seen for 3 time point intensities in Figures 3 and 5, respectively. In each case the spectra comprised of a single spectrum, generated from the top of the peak. Intensities of up to 150,000 counts per ion accumulation are illustrated, where no detector saturation has been observed and mass measurement errors of typically less than 3 ppm have been obtained. The corresponding spectra produced for the remaining parent dextromethorphan are shown in Figure 4. pDRE has enabled exact mass measurement to be obtained routinely over a suitable concentration range for true measurement of metabolite formation and profiling.
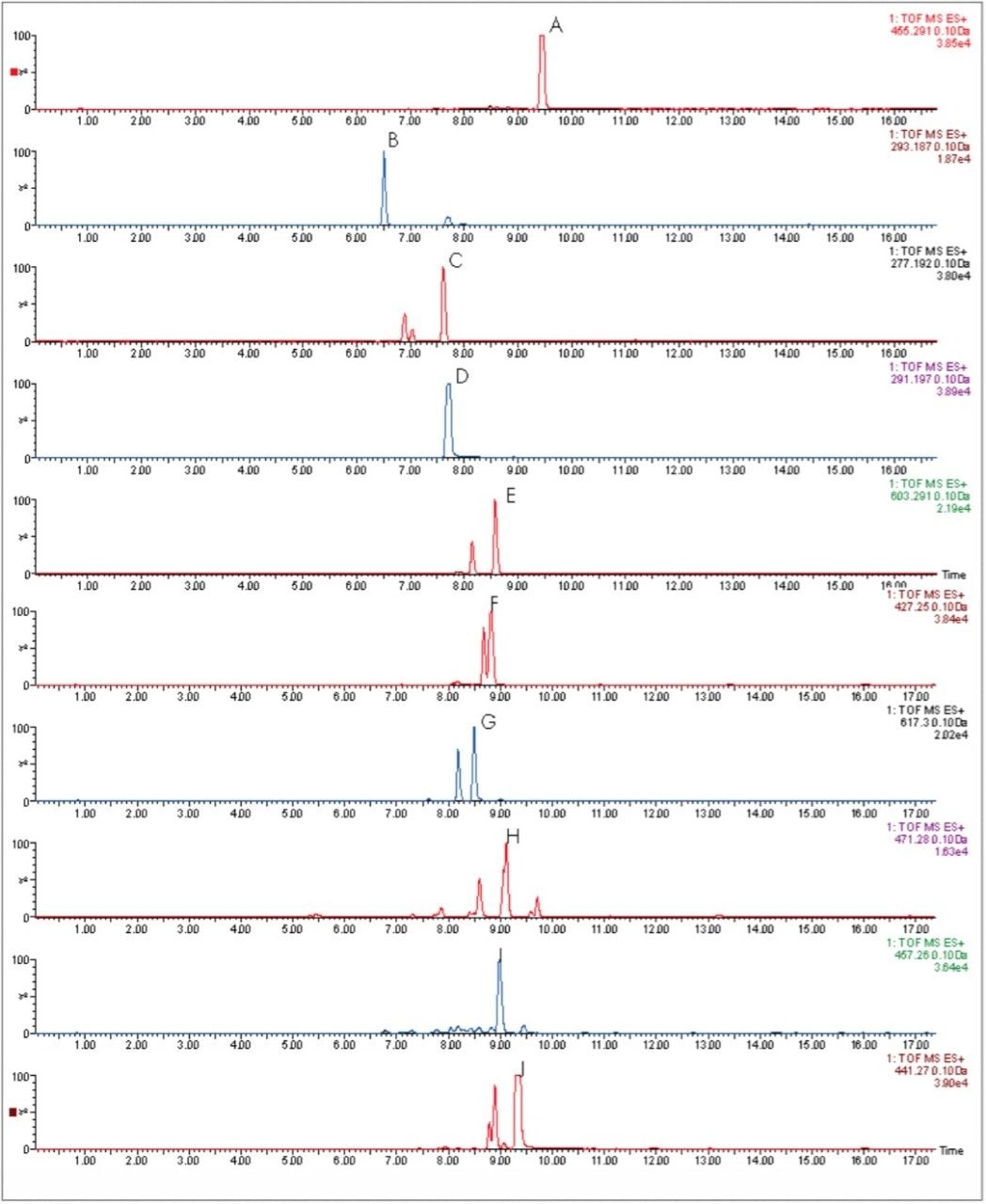
As well as improving mass measurement performance, chromatography technology has also be enhanced by using Ultra Performance LC. In Figures 6 and 7, the HPLC and UPLC-Tof extracted mass chromatograms for metabolites formed for the in vitroincubation of verapamil are presented. The HPLC data was acquired as part of a previous study.1 It is shown that not only is analysis time reduced by greater than 50%, but chromatographic resolution is also improved. Metabolite isomers can now be chromatographically resolved, reducing the risk of “missed” metabolites. Improved chromatographic resolution also adds further opportunity to obtain higher quality MS data, reducing the need for multiple simultaneous MS experiments.
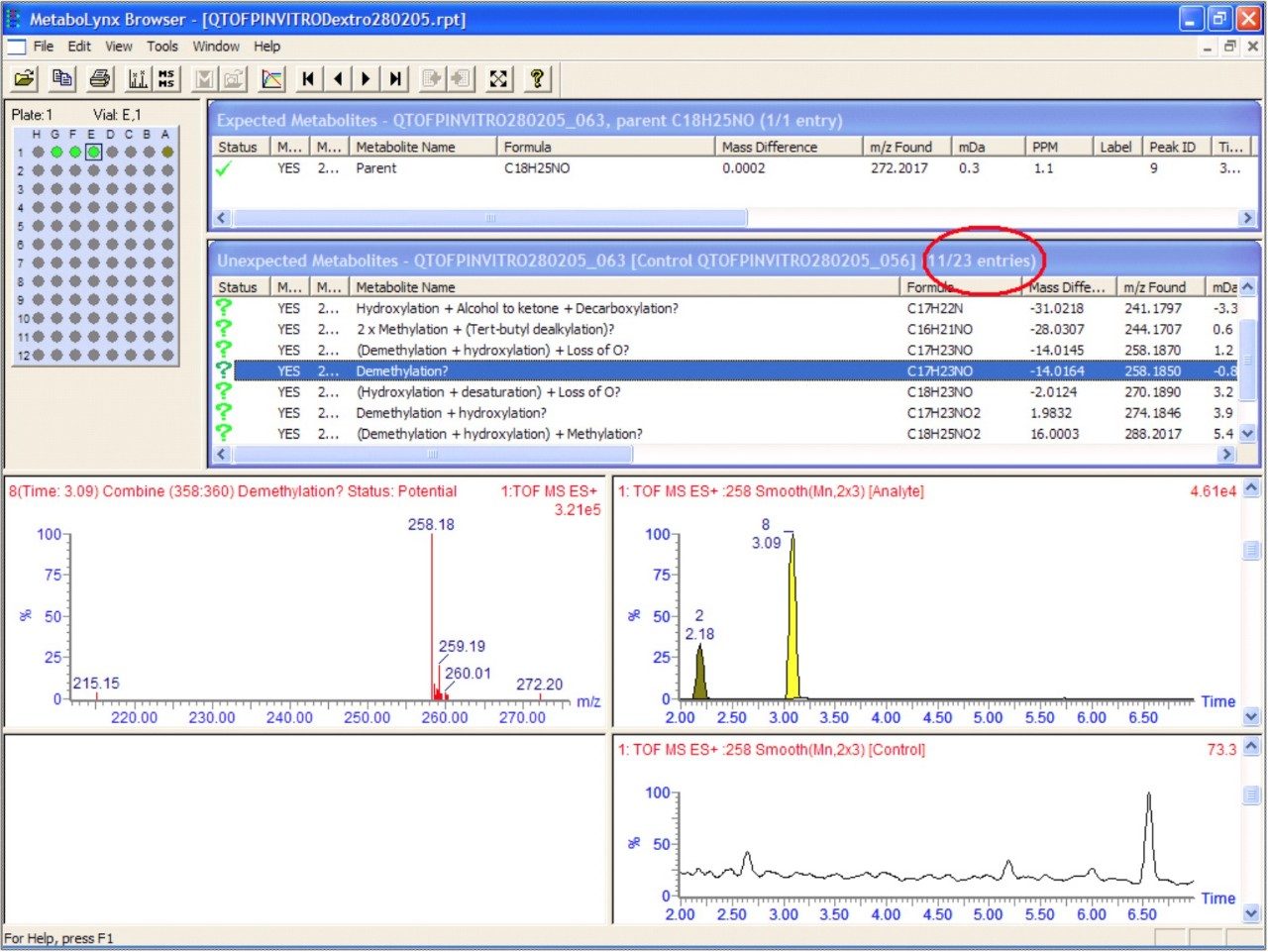
An illustration of the MetaboLynx report browser is contained in Figure 8. The processing of metabolism data is usually a significant bottleneck in metabolite identification. Using MetaboLynx to automatically process the data accelerates this step. The reduced data are displayed on a results browser, which allows rapid interpretation of the data. In addition the data can be greatly simplified by application of the post-processing mass defect data filter. The mass defectproperty of the parent drug will be maintained within a narrow mass range window, for both phase I and phase II biotransformations. The ability to acquire enhanced quality accurate mass spectra provides the opportunity to exploit mass sufficiency and deficiency. Application of a +/–7 mDa mass defect window has enabled the number of unexpected metabolites found to be reduced to 11 from 23. This post-processing step enabled immediate removal of 12 non drug matrix-related unexpected metabolites to be removed.