Perfluoroalkyl substances (PFASs) are a class of anthropogenic compounds that are found in a range of consumer goods and industrial processes due to their chemical properties. Common uses include firefighting foams, insecticide formulations, water-resistant coating, floor polishes, and oilresistant coatings for paper products approved for food contact. Due to their widespread use and subsequent leaching from materials, PFASs are so ubiquitous that they are frequently detected throughout the environment and in 2009, they were classified as persistent organic pollutants (POPs) within the Stockholm Convention.1 Due to their persistent, ubiquitous nature, and possible toxicity, most regulatory agencies worldwide closely monitor the use, occurrence, and impact of both traditional/common and newer, replacement short-chain PFASs.
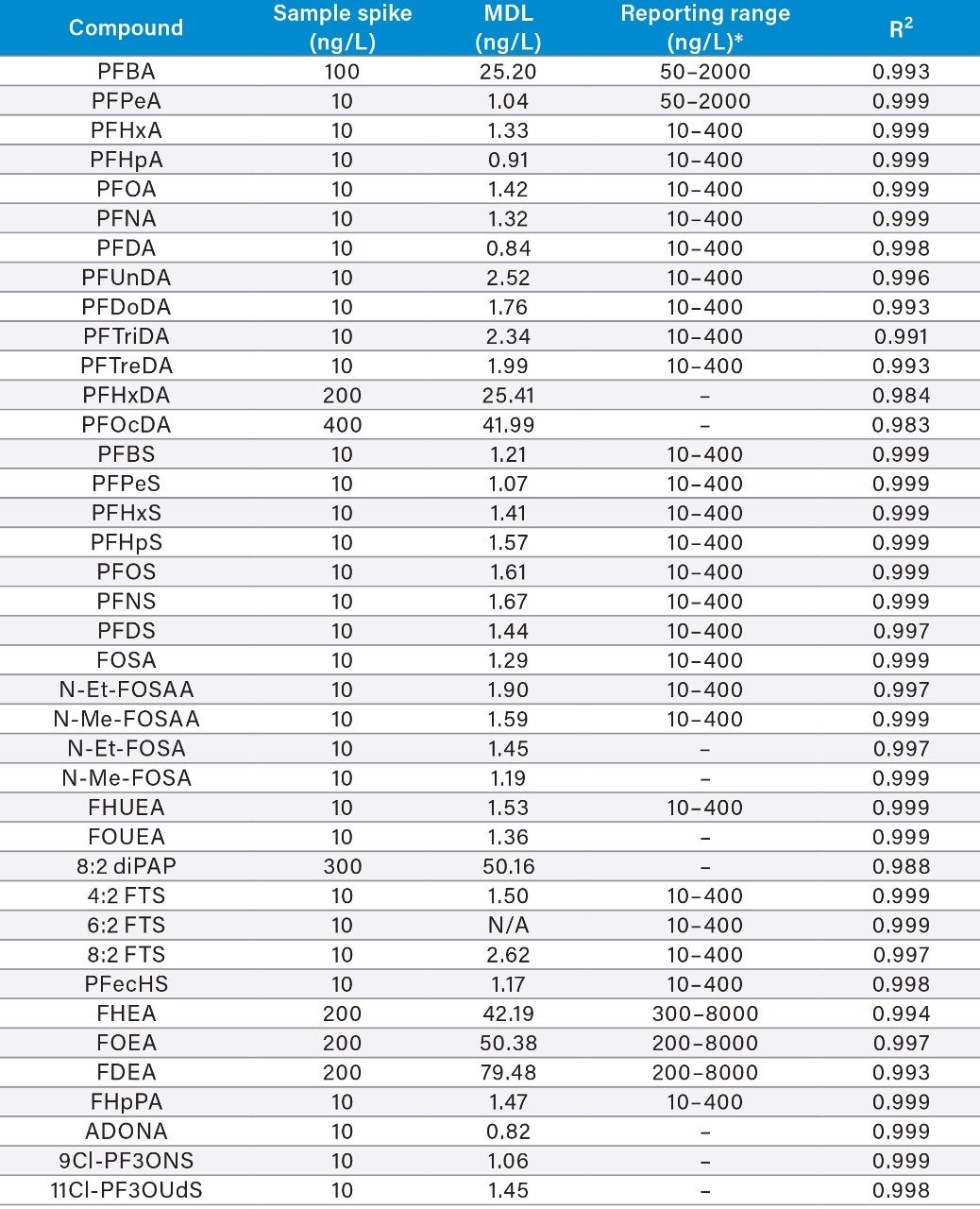
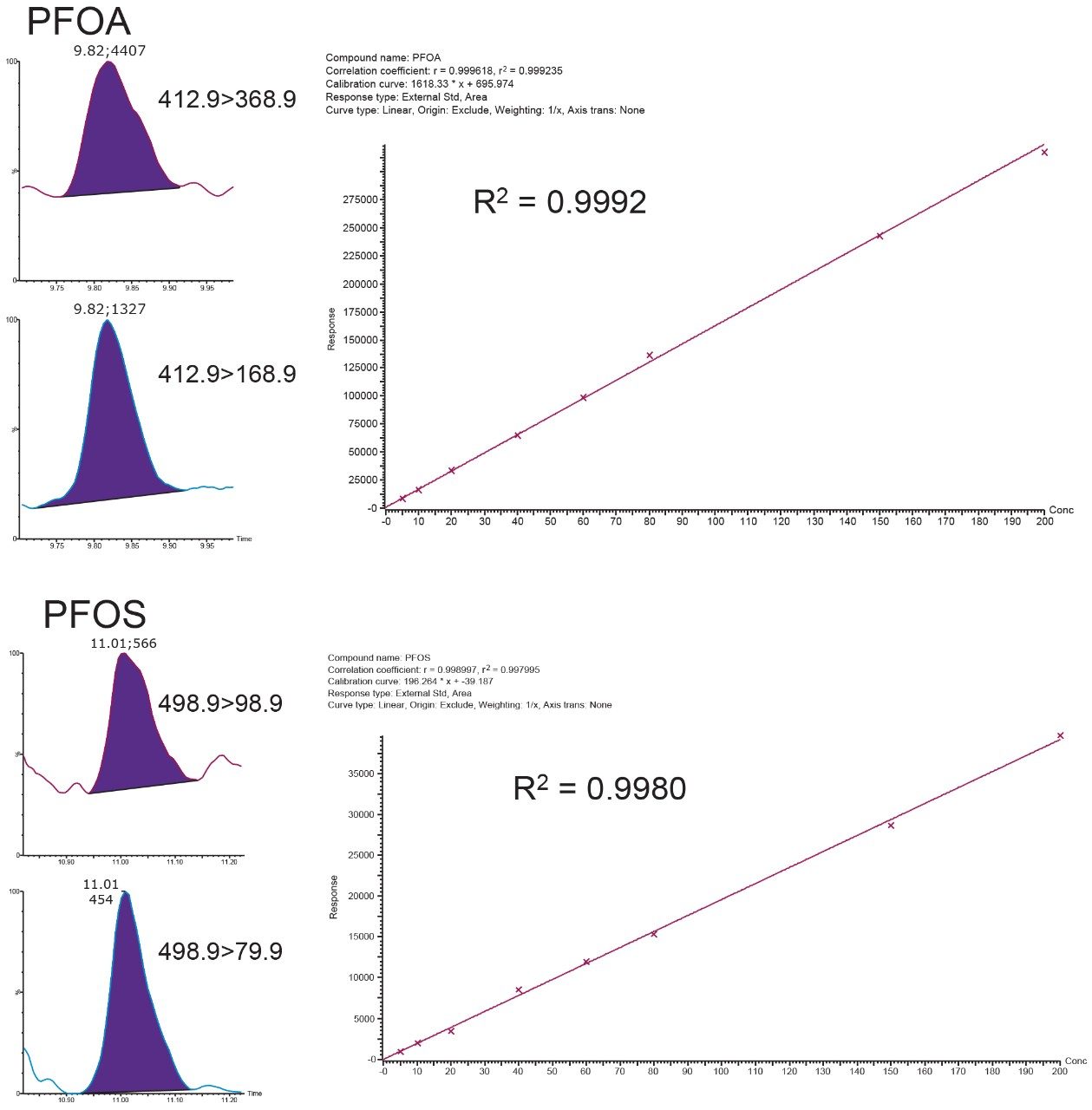
For monitoring and research purposes, ng/L, or part-per-trillion (ppt), detection of PFASs is often required. Within the U.S. drinking water is regulated under the Safe Drinking Water Act, while other environmental waters are regulated under the Clean Water Act. In the third Unregulated Contaminant Monitoring rule (UCMR3)2 for drinking water, the U.S. EPA has required monitoring of six different PFAS compounds with a minimum reporting level in the range of 30 to 200 ng/L for each component. The U.S. EPA has also issued a health advisory3 acute level at 70 ng/L based on the best available peer-reviewed studies of PFAS effects. Within the EU, drinking water is regulated under the Drinking Water Directive, 98/83/EC, while other environmental waters are regulated under the EC Water Framework Directive (WFD), 2013/39/EU.4 In the WFD, PFOS is specifically identified as a “priority hazardous substance.”
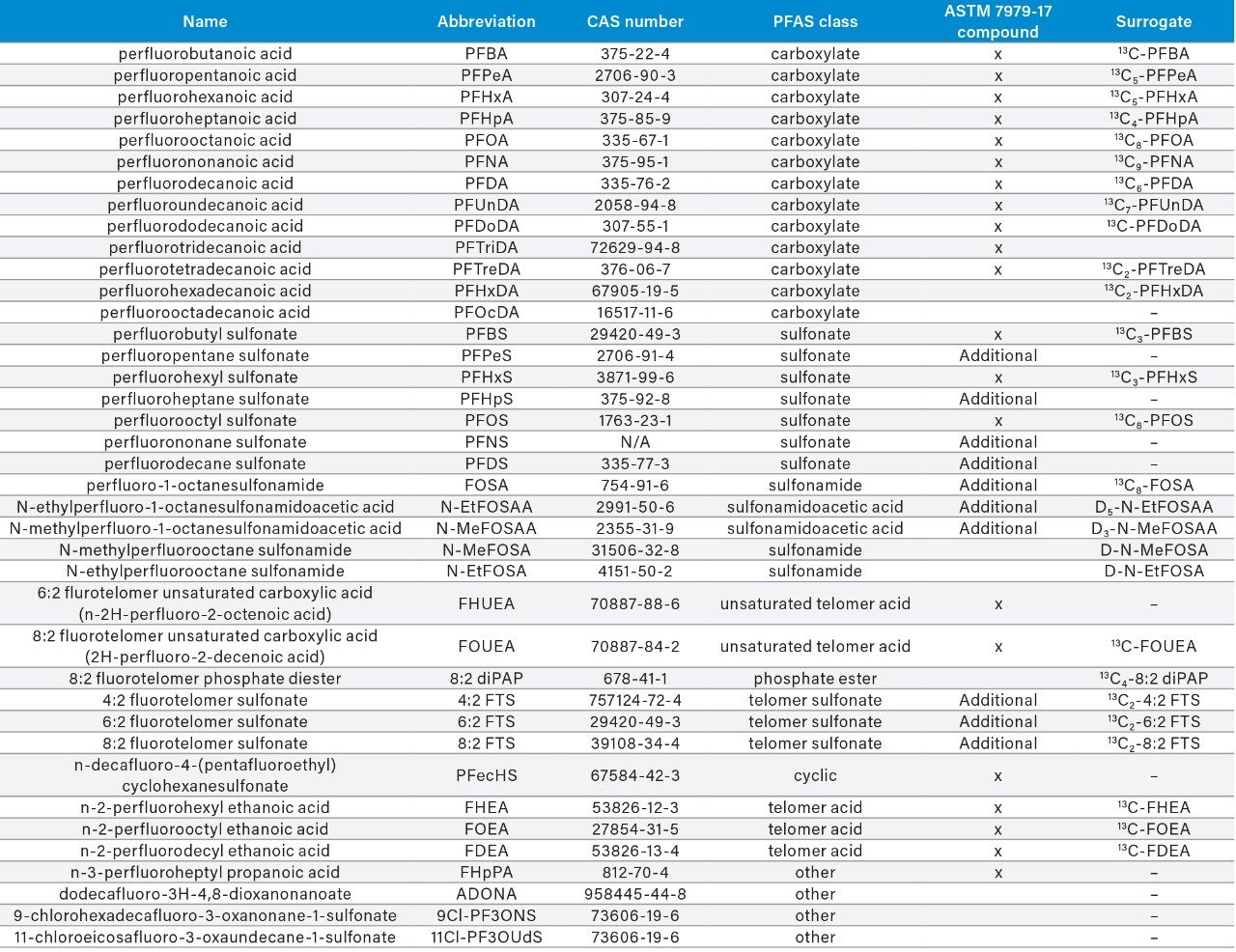
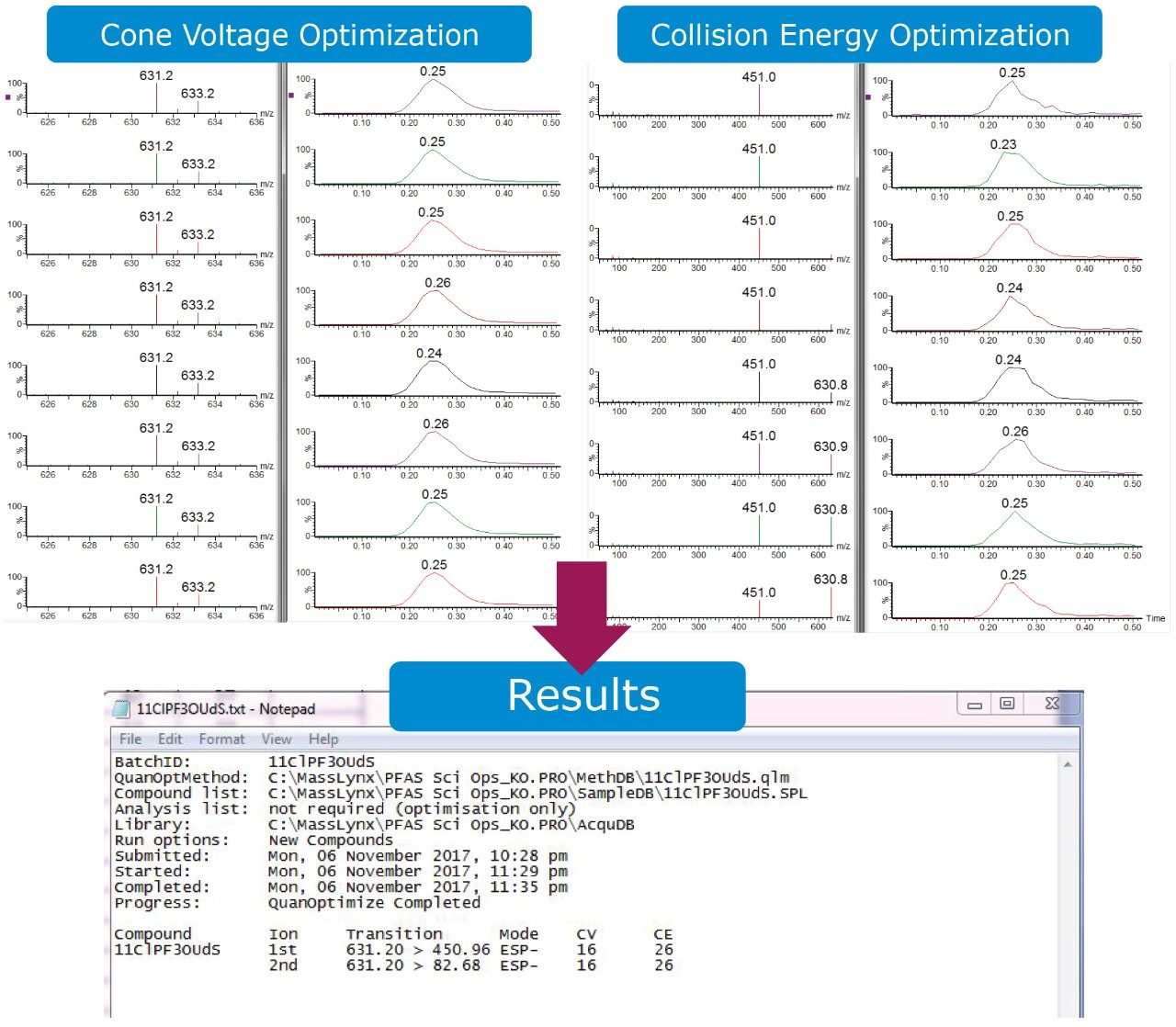
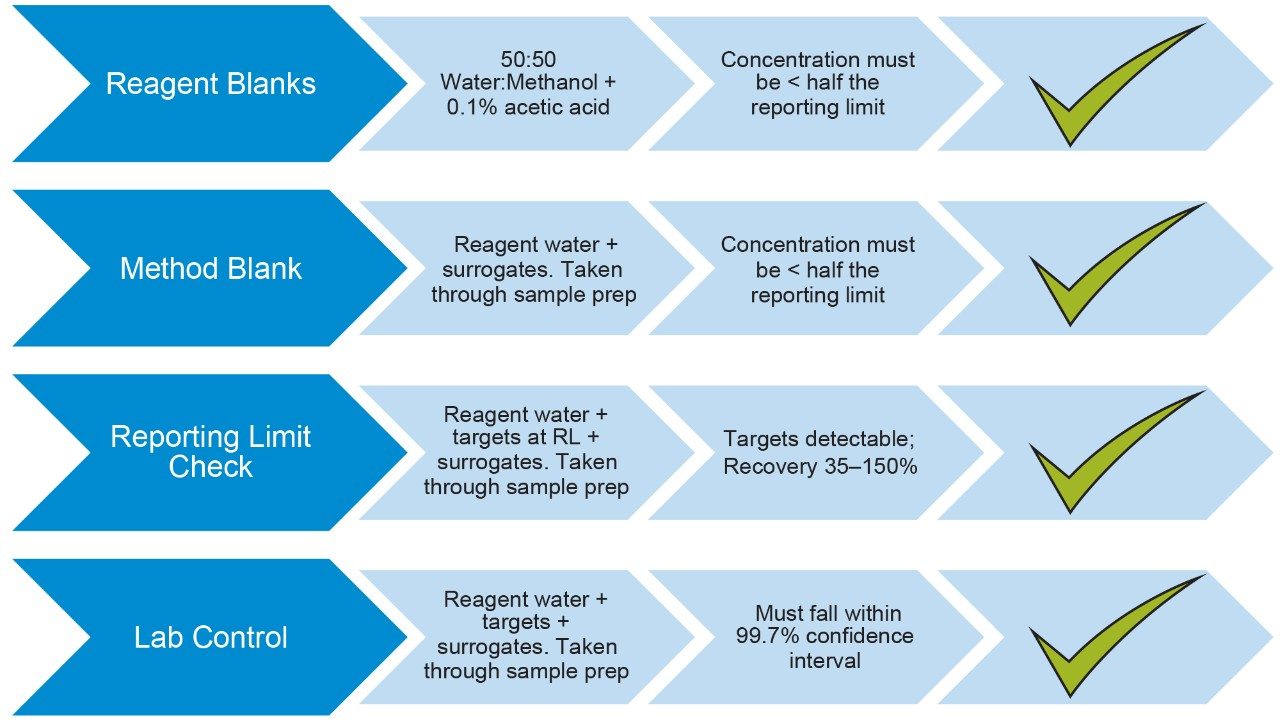
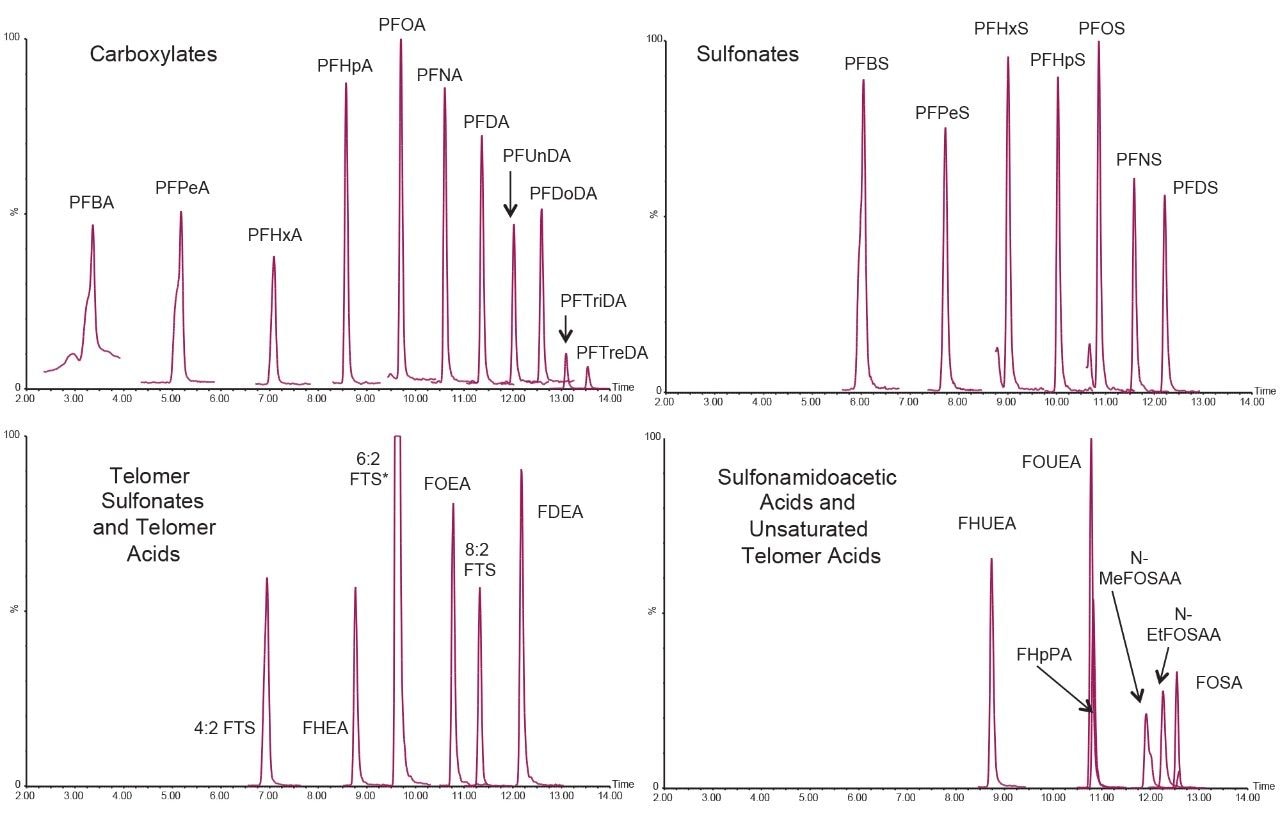
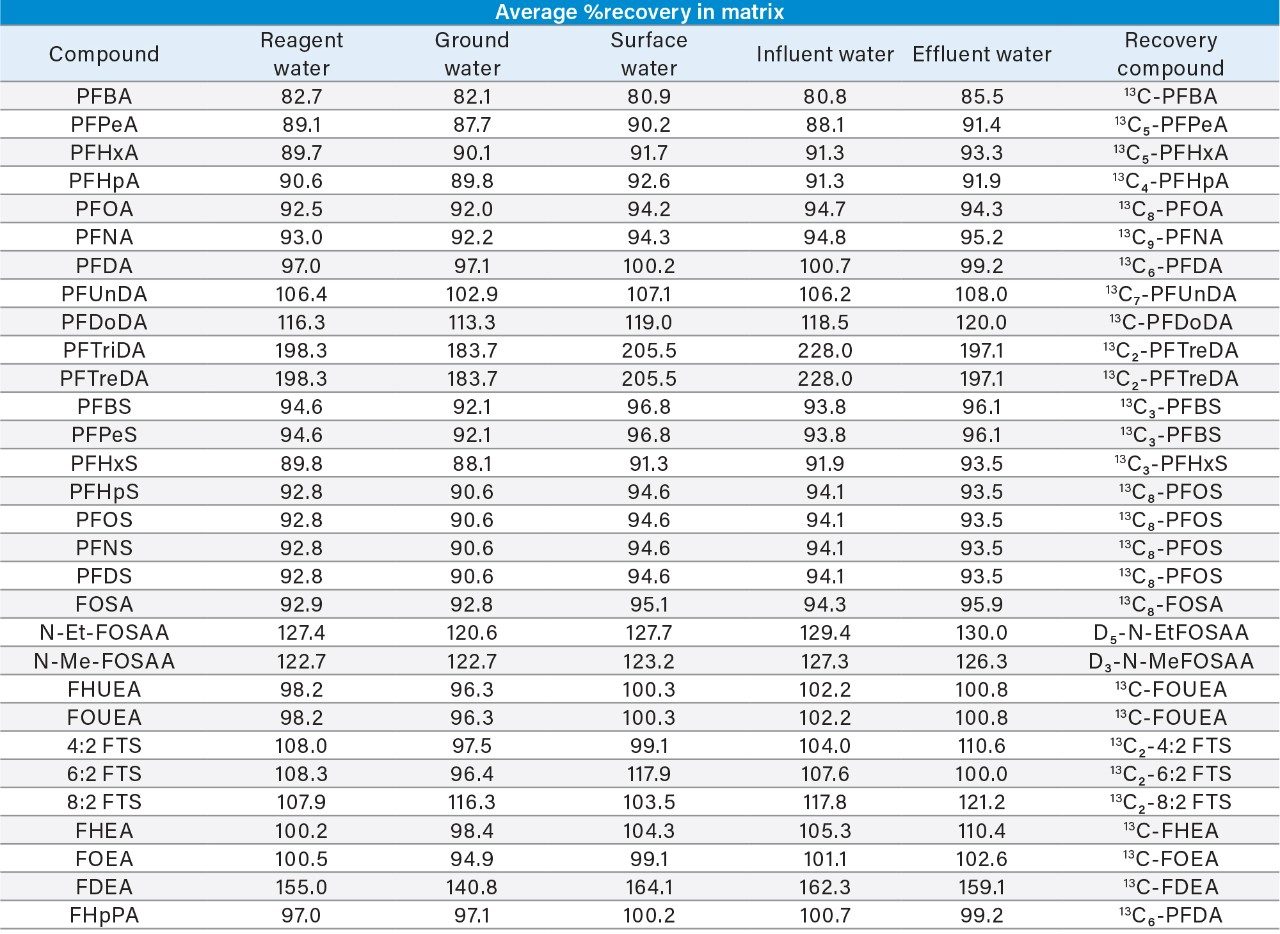
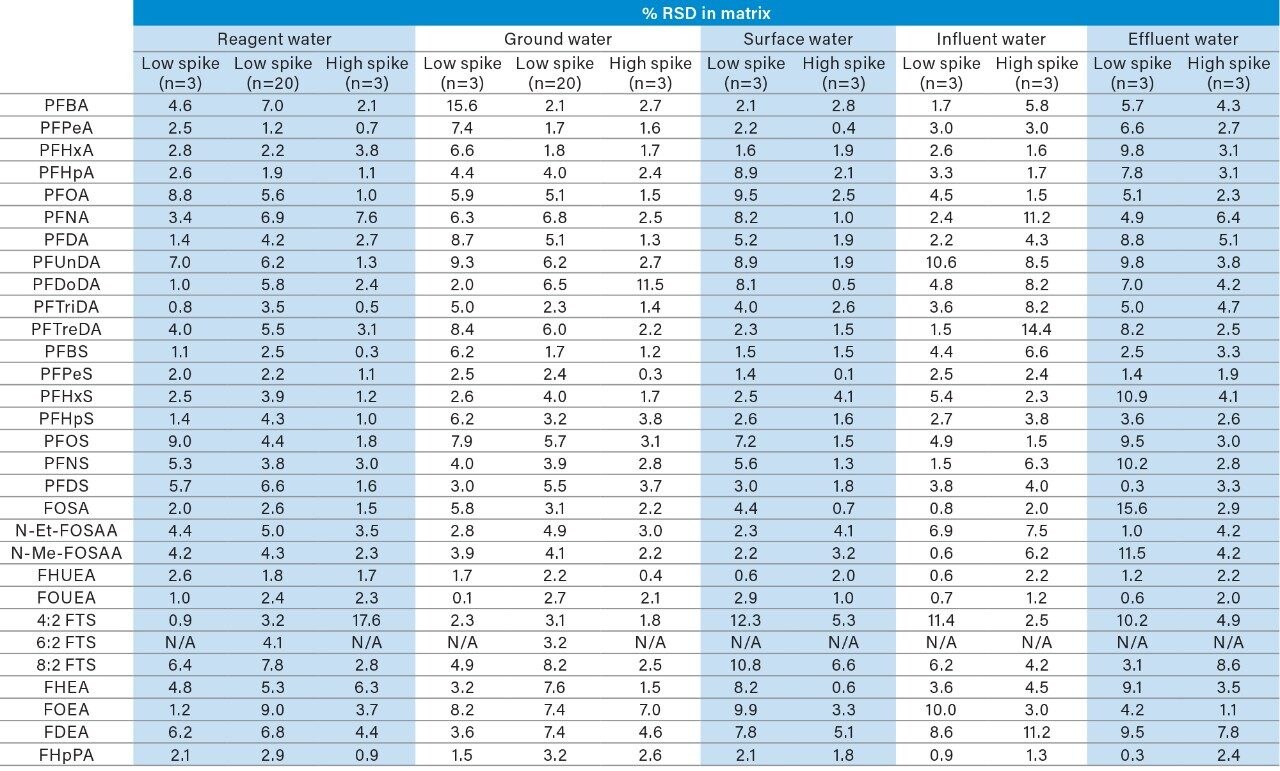
In this application note we describe the use of the recently developed ASTM 7979-17 method (EPA Region 5, Dr. Lawrence B. Zintek)5 to analyze PFASs of interest in environmental waters, not only as described by U.S. legislation, but also those of interest elsewhere, including newer compounds (ADONA, 9Cl-PF3ONS, and 11Cl-PF3OUdS). Since many countries look to the U.S. EPA and other agencies for guidance, it was decided to include as many compounds in a single analysis as was feasible at relevant detection levels.