The removal of weeds from agricultural crops is an important contribution to maintaining the productivity and the quality of those crops. Herbicides are a specific group of plant protection products used to treat a variety of weeds integrated with other control practices such as crop rotation, tillage, and fallow systems. Acidic herbicides comprise families of compounds that include derivatives of benzoic acid (e.g. dicamba), acetic acid (e.g. 2,4-dichlorophenoxyacetic acid [2,4-D]), propanoic acid (e.g. fluazifop), butanoic acid (e.g. 4-(2,4-dichlorophenoxy)butanoic acid [2,4-DB]), picolinic acid (e.g. clopyralid), and other miscellaneous acids such as thiadiazine dioxide (bentazone), and imidazolinones (e.g. imazapyr). Some of the acidic herbicides have been commercially available for almost 80 years and are widely approved for use in agriculture and recreational areas such as golf courses and water courses worldwide. Herbicides can enter water bodies either directly through spray or spray drift, or indirectly via surface water runoff or leaching and sub-surface draining. In the aqueous environment, residues of phenoxyacetic acid herbicides are most commonly found in the form of the free acid.
The presence of pesticides in water is monitored globally, modelled after international norms from the World Health Organization (WHO) which are typically used as the basis for regulations worldwide.1 Presence of pesticides in European waters is regulated through different directives. The EU Drinking Water Directive sets a maximum limit of 0.1 μg/L for individual pesticide residues present in a sample (0.5 μg/L for total pesticides).2 The Water Framework Directive (WFD) deals with surface waters, coastal waters, and groundwater.3 Member States must identify River Basin Specific Pollutants and set their own national environmental quality standards (EQSs) for substances that may have a harmful effect on biological quality and have been identified as being discharged into the water environment in significant quantities. Values for these EQS vary across Europe; for example, the annual average EQS for 2,4-dichlorophenoxyacetic acid (2,4-D) was reported as 0.1 μg/L in France and Germany but 26 μg/L in the Netherlands.4 In the USA, drinking water is regulated under the Safe Drinking Water Act, where there are varying maximum contaminate levels for each residue.5
Other environmental waters are regulated under the Clean Water Act.6 Although regulations vary from country to country, many look to guidelines established by the WHO, EU, or USA.
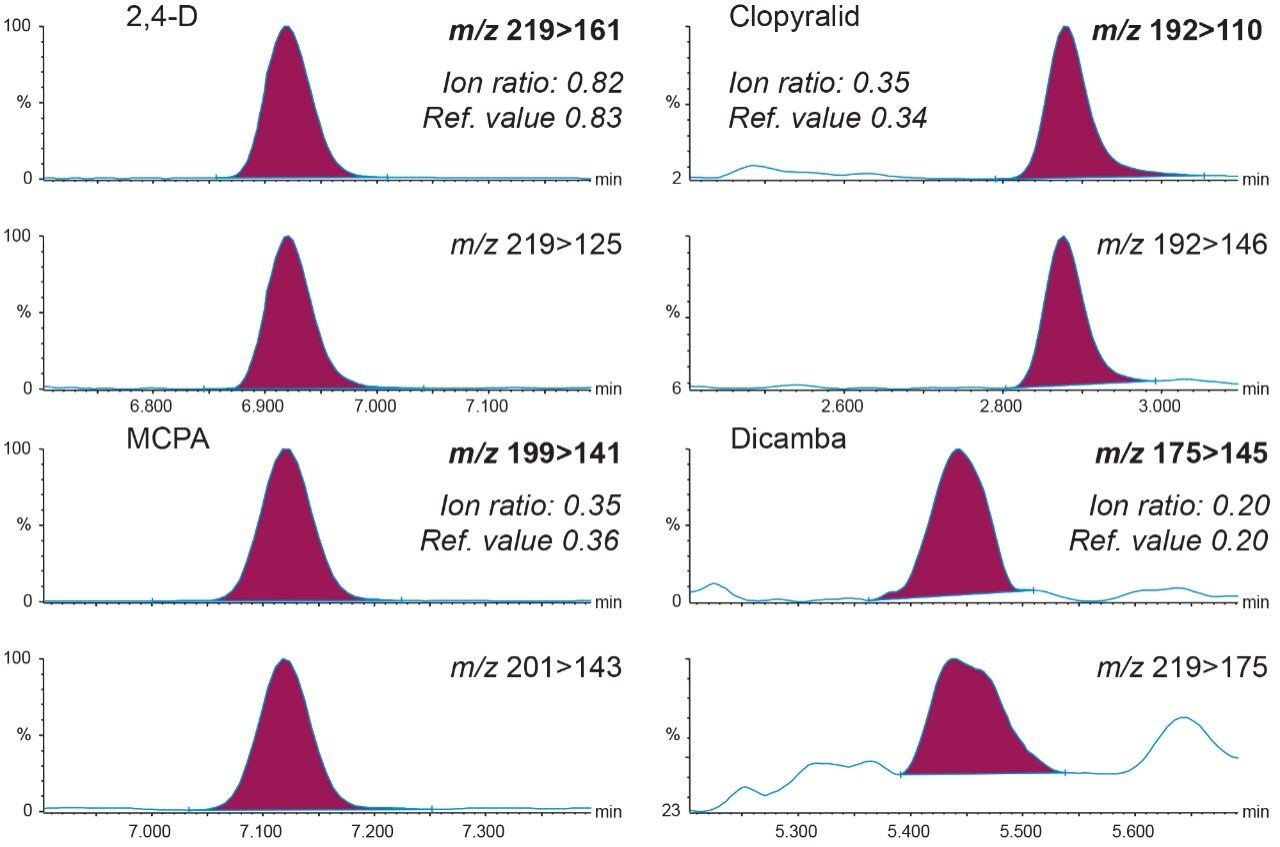
There is a need for reliable analytical methods for monitoring acidic herbicides in drinking and surface waters. Historically used methods are liquid-liquid extraction or solid-phase extraction (SPE) as a concentration step. Liquid chromatography with tandem mass spectrometry (LC-MS/MS) has now supplanted gas chromatography-mass spectrometry (GC-MS) due to ease of use and no need or minimal sample preparation.
This application note describes a rapid method for the determination of a range of acidic herbicides in water samples using large volume direct injection by LC-MS/MS on Waters ACQUITY UPLC I-Class System coupled to the Xevo TQ-XS to achieve best degree of performance and ultra-high sensitivity.