A tryptic digest of an Escherichia coli (E.coli) lysate was spiked with TMT labeled bovine serum albumin (BSA) at various concentrations. The peptides were separated and analyzed using a nanoACQUITY UPLC System coupled to a SYNAPT G2 HDMS. The data were acquired in LC-HDMSE mode – an unbiased mobility assisted TOF acquisition method – switching between low and elevated energy on alternate scans.
Subsequent correlation of precursor and product ions can then be achieved using both retention and drift time alignment. Searches were conducted with ProteinLynx GlobalSERVER using a species specific database to which sequence information of BSA and other contaminating species was appended. Searching the DB with TMT as a fixed modification was expected to only identify BSA, whereas variable searches should identify both E.coli proteins and BSA. Moreover, acquisitions that were not supported by means of ion mobility separations are expected to produce fragment E.coli ion spectra comprising the TMT reporter ions of co-eluting BSA peptides.
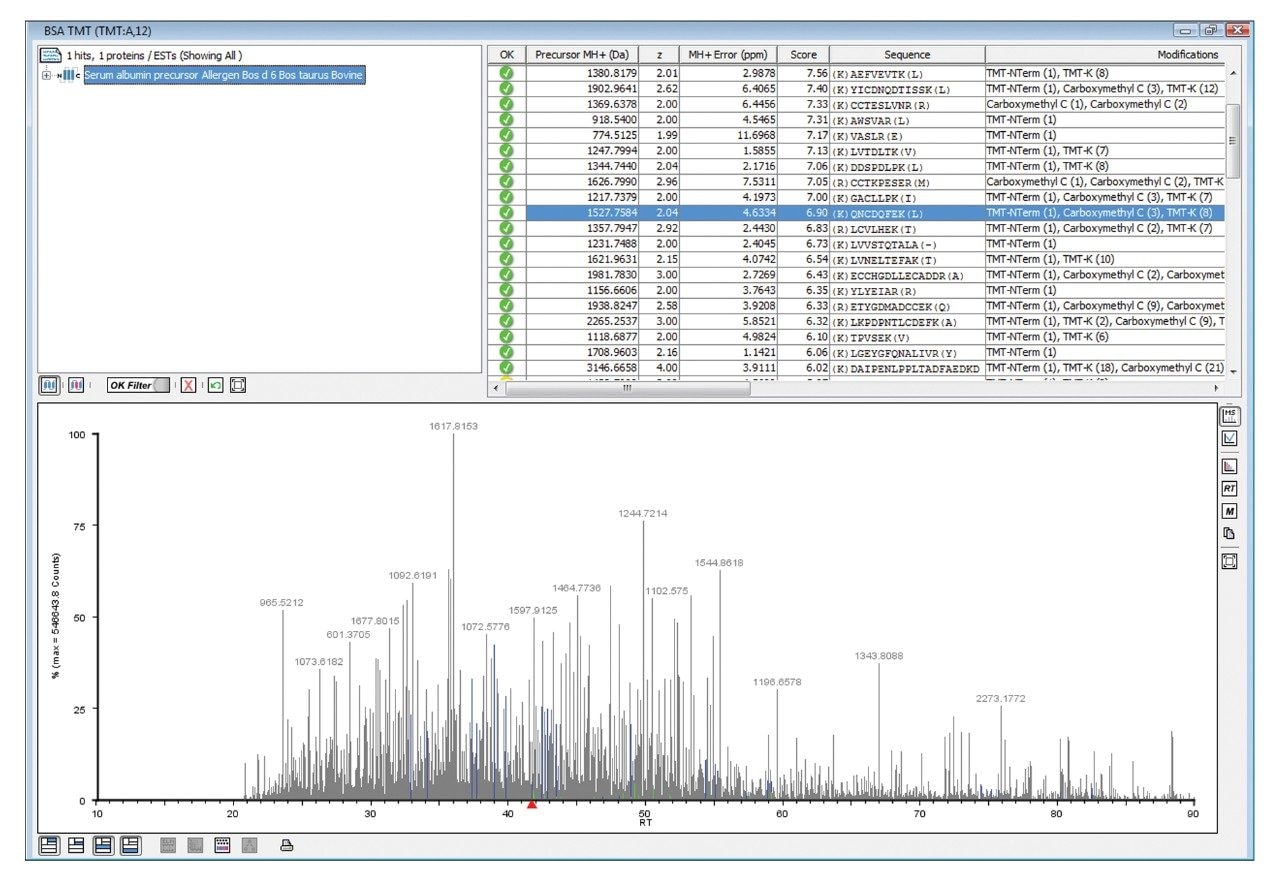
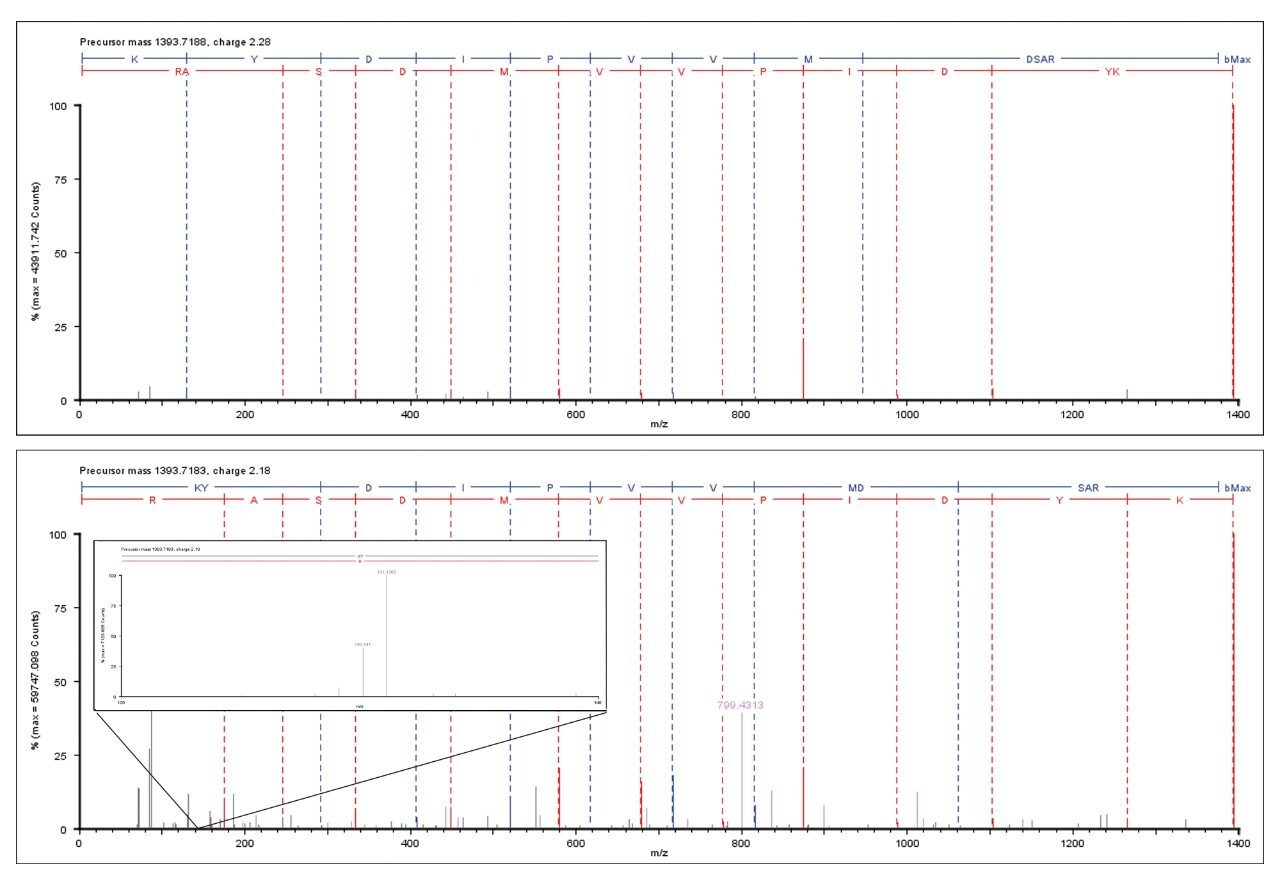
Figure 1 illustrates the identification overview for an HDMSE acquisition with the TMT modification set as a fixed modification for a condition combined amount of approximately 70 fmol labeled BSA added to 750 ng E.coli. In contrast the search results of the same data with TMT specified as a variable modification resulted in 434 E.coli protein identifications, confirming the expected specificity afforded by the acquisition method and applied search algorithm. The results shown in Figure 2 demonstrate that without mobility separation, E.coli fragment ion spectra can be incorrectly appended with TMT reporter ions originating from co-eluting labeled BSA peptides, illustrating the requirement for increased system peak capacity via orthogonal peptide separation. Moreover, the spectrum shown in the bottom pane of Figure 2 comprises other contaminating product ions that do not arise from the precursor of interest and could have lead to an incorrect assignment if the multiplexed nature of the data would not have been acknowledged by the deconvoluting and search algorithms.