Metabolite identification is an integral part of discovery and development programs within pharmaceutical companies of any size. One of its most significant bottlenecks is the structural elucidation process. Traditionally this process has been conducted with nominal mass instrumentation such as tandem quadrupole and ion trap mass spectrometers, with time-consuming manual interpretation of the spectral data. In recent years, there has been a shift to the use of platforms that are capable of high resolution and exact mass measurement (e.g., time-of-flight (Tof) or quadrupole time-of-flight (QTof) mass spectrometry), since their additional levels of specificity reduce falsepositive identifications. Such technologies also improve the process of characterizing complex fragmentation pathways.
The SYNAPT G2 HDMS System delivers a paradigm shift for the metabolite identification process due its ability to combine the full power of UPLC separations,1 high-resolution mass spectrometry, and intelligent informatics to significantly reduce or eliminate false positive results in the structural elucidation process.
SYNAPT G2 HDMS, with its breakthrough quantitative Tof technology, QuanTof, provides up to 40,000 FWHM, sub-1-ppm RMS mass accuracy, enhanced mass precision, and enhanced dynamic range (>104) – all at acquisition rates of up to 20 spectra/sec. This is unlike FT-MS or electrostatic ion trap-based MS systems whose resolution and dynamic range reduces significantly as spectral acquisition rate and sample complexity respectively increase.
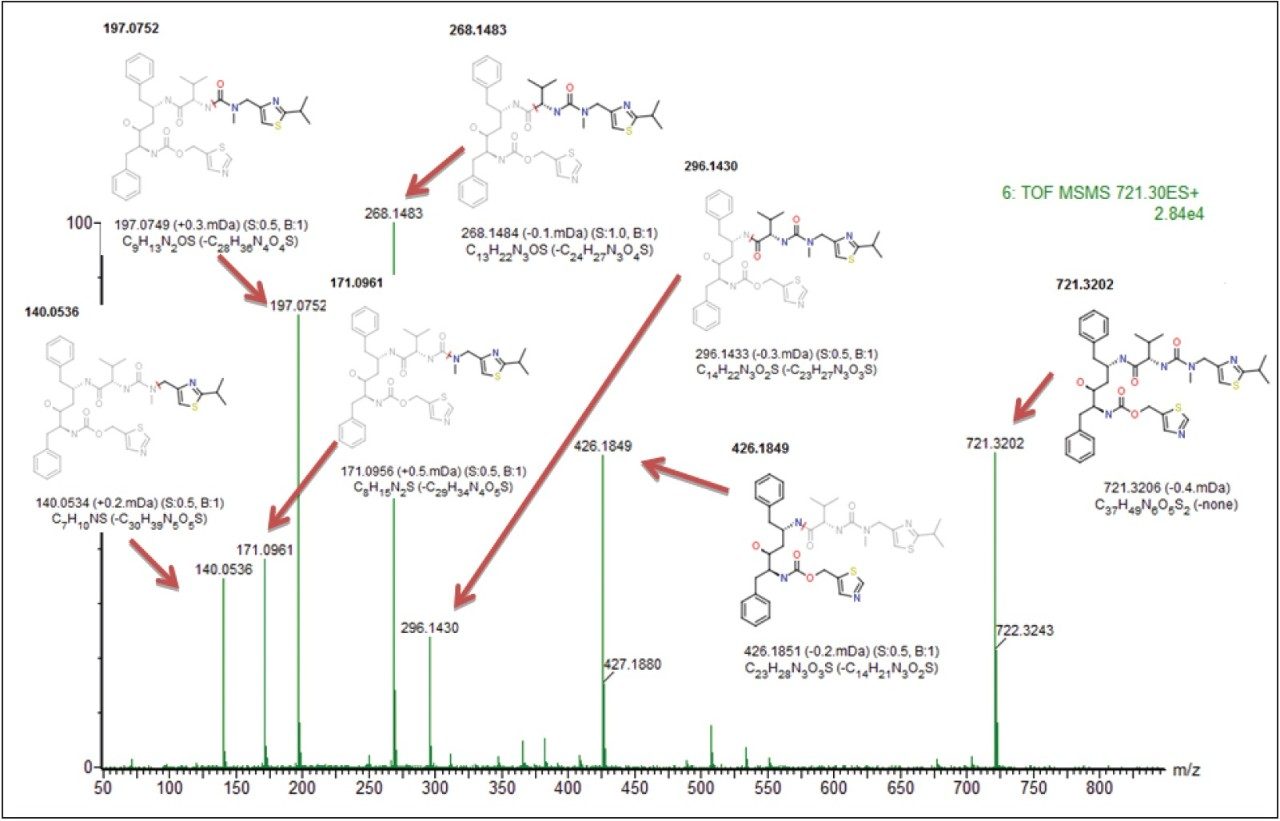
SYNAPT G2 HDMS collects comprehensive fragment ion (MS/MS) information from every detectable molecular ion across the entire chromatographic separation provided by UPLC.
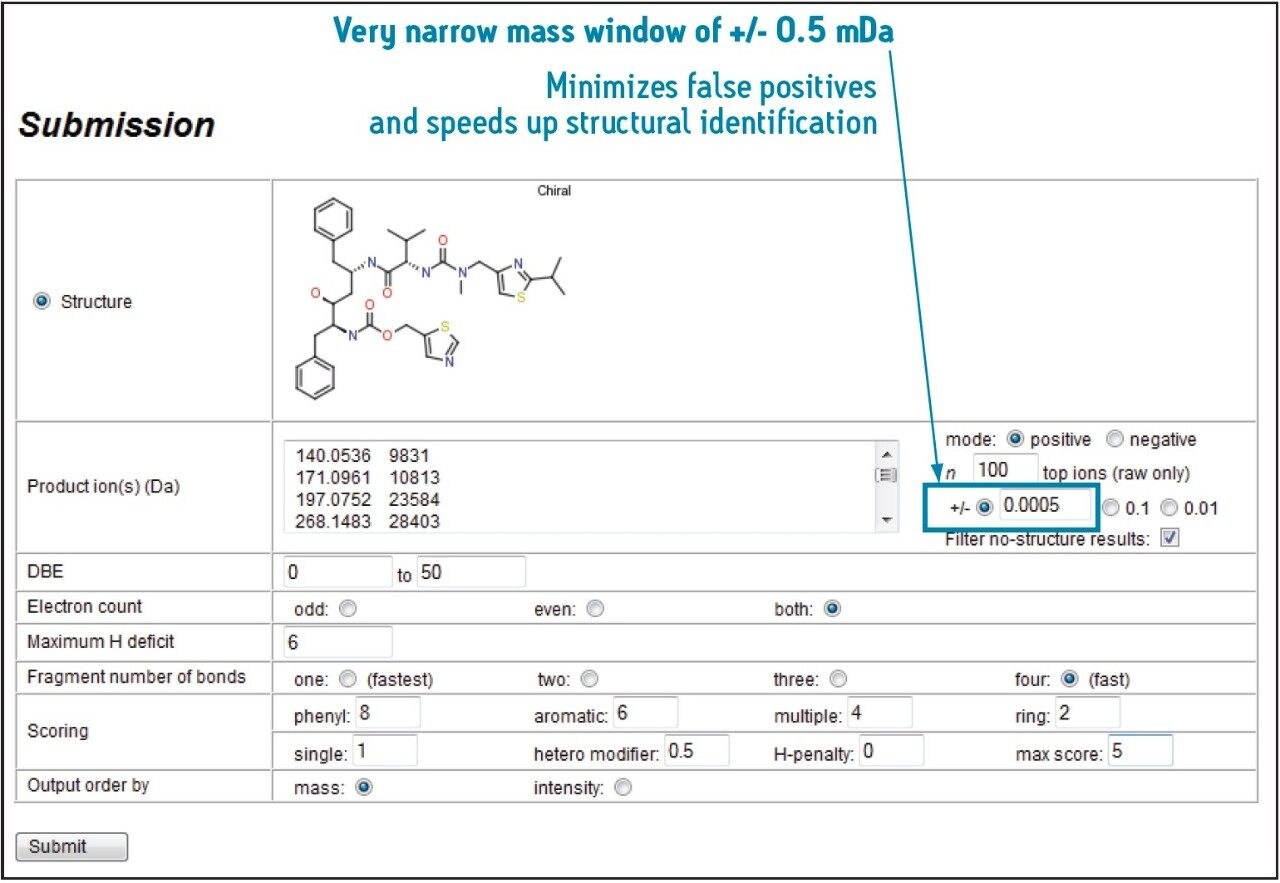
MassFragment,2 an intelligent software tool, automates structural assignment to fragment ion spectra, making data processing significantly easier.